

The Customer
The client is a leading pharmaceutical company committed to maintaining the highest standards of quality, compliance, and safety in its manufacturing and operations. They aim to enhance efficiency while ensuring strict adherence to regulatory guidelines, particularly in the management of computerized systems.
The Project
The project focused on implementing and validating Computerized System Validation (CSV) procedures for the client’s critical systems. These systems play a vital role in managing production data, ensuring compliance with regulatory bodies, and maintaining product safety and efficacy.
Industry 4.0 – Bridging the Analog and Digital Worlds
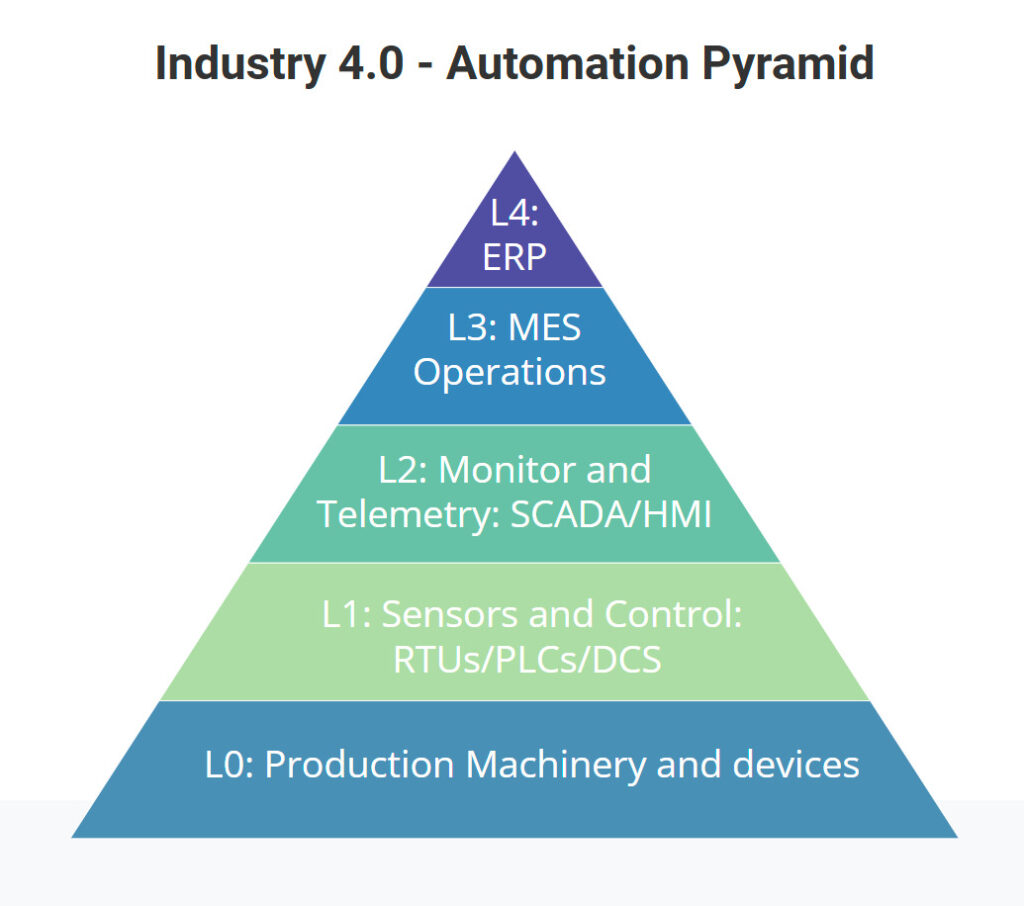
From the ancient times of primitive exploration and exploitation of natural resources for the advance of humanity up to the modern practices of production and manufacturing, we’ve come up to a point where everything is digital! Including the factories, as per the following illustration.
V-model diagram of MES software implementation
Validation plan
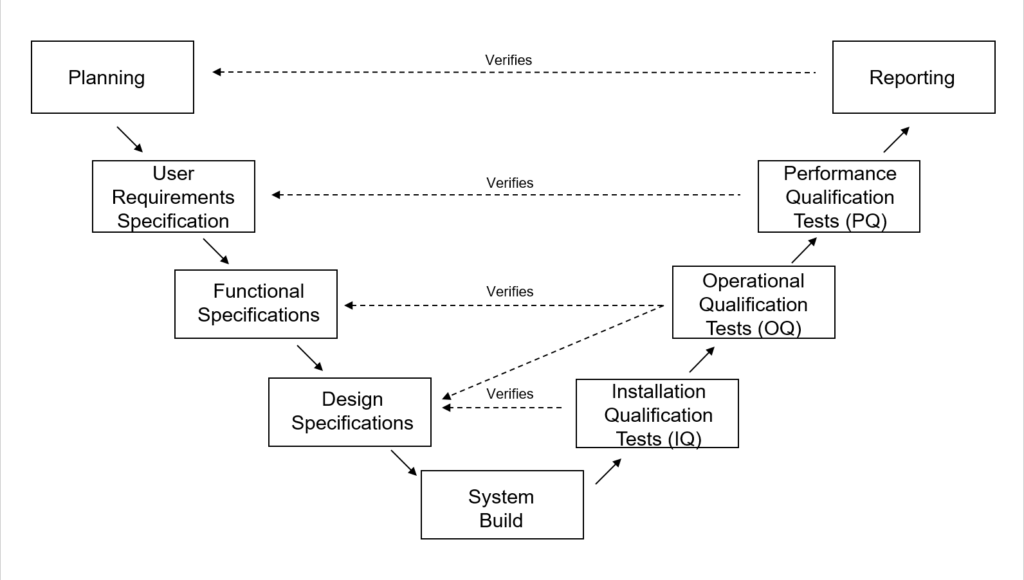
The Validation Plan defines what will be validated and the approach you will use. It also defines roles and responsibilities along with the most important part, the Acceptance Criteria.
User Requirements Specification (URS)
The User Requirements Specification describes what the user needs from the software and how they will use it. It also contains any critical constraints such as regulations, safety requirements, operational requirements, etc.
For example, here’s a list of a few User Requirements that might be needed for a lab system.
- System must track training of Production and Quality Assurance laboratories analysts on laboratory methods and techniques.
- System must track samples coming into the laboratories.
- System must automatically assign lab analysts to test samples based on availability and training.
- System must send sample testing status outcomes to the ERP level.
- System must comply with the relevant legislatives and regulatory procedures provided by authority bodies and agencies.
Functional Specifications (FS)
The Functional Specification document describes how the software needs to work and look to meet the user needs. The document might include descriptions of how specific screens and reports should look or describe data that needs to be captured.
The Functional Requirements can also include logic and calculations along with how it will comply with regulatory requirements. For example, the Part 11 compliance requirements might detail how passwords or the audit trail should work.
Design Specifications (DS)
- The Design Specification document is one that contains all of the technical elements of the software or systems. This includes:
Database Design – file structures, data structures, field definitions, data flow diagrams, entity relationships. - Logic or Process Design – pseudo code for logic and calculations.
- Security Design – protection from malicious software.
- Interface Design – what data will move from one system to another, how and how often.
- Architecture Design – required hardware, operating systems, application versions, middleware, networks, protocols.
- Specific peripheral devices – scanners, printers, digital laboratory equipment, metrology devices.
System Build
In the System Build step, you develop or purchase your software and then configure it to the previous specification documents. This step includes unit testing and integration testing.
Installation Qualification Tests (IQ) Tests
The Installation Qualification tests provide confirmation that the software or system is installed and setup according to the Design Specification.
Usually the software is first installed in a test or validation environment, but there can be exceptions in situations such as manufacturing.
Operational Qualification (OQ) Tests
Operational Qualification testing is often referred to as Functional Testing or System Testing. OQ tests confirm that all functionality defined in the Functional Specification is present and working correctly, and that there are no bugs.
OQ tests can also include confirmation of any design elements not tested during IQ, such as configuration, are working as specified.
Performance Qualification (PQ) Tests
Performance Qualification testing is often called User Acceptance testing. PQ testing confirms that the software will meet the users’ needs and is suitable for their intended use, as defined in the User Requirements Specification.
Testing can follow Use Cases, SOPs and/or user-defined scenarios. For simple software like reports or spreadsheets, OQ and PQ testing are often combined.
Reporting
The last step in this validation method is to write the Validation Report, often called the Validation Summary or System Certification.
This report provides confirmation that all activities specified in the validation plan have been completed.
The Validation Report summarizes the testing results and provides confirmation that all acceptance have been met and the software is ready for deployment.
Ensuring Data Integrity
In the pharmaceutical industry, accurate data is not just a requirement but a cornerstone of safety and efficacy.
CSV procedures establish robust mechanisms to validate computerized systems that manage, process, or store critical data.
This validation ensures that data remains accurate and consistent throughout its lifecycle, from creation to archival.
By meticulously verifying these systems, pharmaceutical companies mitigate the risk of data corruption, unauthorized access, or loss, thereby upholding the integrity of their operations and regulatory obligations.
Compliance with Regulatory Standards
Compliance with stringent regulatory standards such as Good Manufacturing Practices (GMP) and Good Clinical Practices (GCP) is non-negotiable in the pharmaceutical industry.
CSV procedures serve as a vital tool in achieving and maintaining compliance with these standards. They provide documented evidence that computerized systems operate reliably and consistently within predefined specifications.
This documentation is essential during regulatory inspections, demonstrating adherence to guidelines and ensuring that products meet the highest quality and safety standards provided by the leading worldwide bodies like US FDA, EMA, WHO, ICH, CFDA, ISPE.
Enhancing Operational Efficiency
Efficiency and productivity are key drivers in pharmaceutical manufacturing. CSV procedures streamline processes by identifying potential issues within computerized systems before they impact operations.
By conducting thorough validation protocols, companies can proactively address vulnerabilities, optimize system performance, and minimize downtime.
This proactive approach not only enhances operational efficiency but also supports continuous improvement initiatives, ensuring that pharmaceutical companies remain agile and res

Risk Mitigation and Patient Safety
Ultimately, the primary focus of CSV procedures in the pharmaceutical industry is to mitigate risks and enhance patient safety.
By validating computerized systems, companies reduce the likelihood of errors in data processing or reporting, which could have serious implications for patient health.
Whether it’s ensuring accurate clinical trial data or maintaining consistent manufacturing processes, CSV procedures play a pivotal role in safeguarding the reliability and efficacy of pharmaceutical products.
Future Directions in CSV
As technology evolves, so too must CSV procedures. The advent of advanced analytics, artificial intelligence, and big data in pharmaceuticals necessitates robust validation frameworks that can adapt to these innovations.
Future-proofing CSV procedures involves incorporating flexible validation methodologies and staying abreast of emerging technologies to maintain industry leadership and regulatory compliance.
In conclusion, CSV procedures are not just a regulatory requirement but a cornerstone of quality assurance and operational excellence in the pharmaceutical industry.
By rigorously validating computerized systems, companies uphold data integrity, ensure compliance with regulatory standards, enhance operational efficiency, mitigate risks, and ultimately safeguard patient safety.
As the industry continues to evolve, embracing innovative approaches to CSV will be essential in driving continuous improvement and maintaining the highest standards of pharmaceutical excellence.
Overview
With the increasing reliance on digital systems in pharmaceuticals, the client needed a comprehensive approach to ensure their systems’ reliability, accuracy, and compliance.
The goal was to implement CSV procedures that would ensure data integrity, system performance, and adherence to industry regulations like GMP and GCP.
The Solution
Our team worked closely with the client to develop and implement a CSV strategy tailored to their specific needs. This included drafting validation plans, creating user requirement specifications (URS), functional specifications (FS), and design specifications (DS). We executed thorough testing protocols, including Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ), to validate the computerized systems.
The Business Benefits
- By implementing CSV procedures, the client achieved:
Regulatory Compliance: Ensured that all computerized systems met regulatory requirements. - Improved Data Integrity: Safeguarded the accuracy and consistency of critical data throughout the system lifecycle.
- Operational Efficiency: Enhanced system performance, reducing the risk of downtime and ensuring smooth operations.
- Risk Mitigation: Reduced the potential for data errors and system failures, thereby enhancing patient safety and product reliability.